Métodos de combinación de p-values para detectar una asociación incompleta
Los metanálisis aumentan el poder estadístico al combinar los resultados de pruebas estadísticas de múltiples estudios. Los métodos de metanálisis se han evaluado principalmente con la condición de que todos los datos de cada estudio tengan una asociación con el fenotipo dado. Sin embargo, las condiciones experimentales específicas en cada estudio o la heterogeneidad genética pueden resultar en “estadísticas no asociadas” que se derivan de la distribución nula.
En este artículo muestran que el poder de los métodos de metanálisis convencionales disminuyen rápidamente a medida que se incluye un número creciente de estadísticas no asociadas, mientras que el método clásico de Fisher y su variante ponderada (wFisher) exhiben un poder relativamente alto que es robusto a la adición de estadísticas no asociadas.
También proponen otro método basado en la distribución conjunta de p-values ordenados (ordmeta). Los análisis de simulación para la prueba t, la secuencia de ARN y los datos de microarreglos demostraron que wFisher y ordmeta superaron a los métodos de metanálisis existentes cuando solo un pequeño número de estudios tienen una asociación.
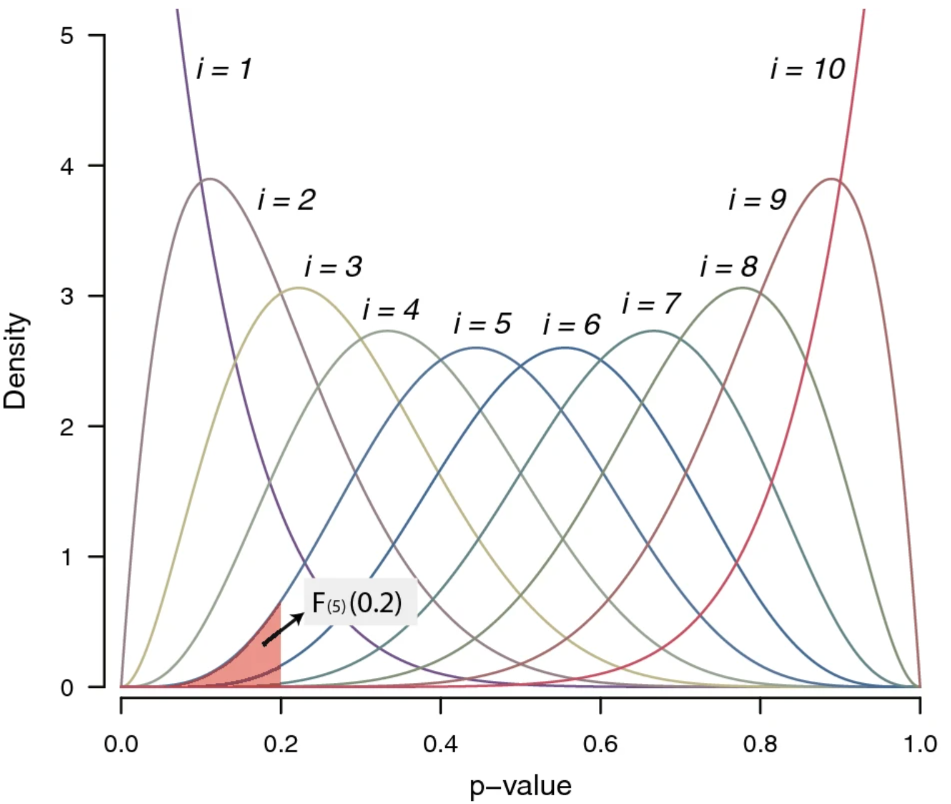
Finalmente realizaron metanálisis de nueve conjuntos de datos de microarreglos (cáncer de próstata) y cuatro conjuntos de datos de resumen de asociación (índice de masa corporal), donde estos métodos exhibieron una alta relevancia biológica y fueron capaces de detectar genes que los métodos más avanzados pasaron por alto.
Los métodos de combinación de p-values son generalmente útiles cuando se combinan datos heterogéneos independientemente de los modelos de datos y los métodos estadísticos aplicados en cada estudio. Estos métodos pueden proporcionar muchos hallazgos nuevos en metanálisis de expresión génica, GWAS y otros datos genómicos y médicos.
Lee más:
https://www.nature.com/articles/s41598-021-86465-y