

¿Qué es el llamado de variantes?
El llamado de variantes es el proceso mediante el cual identificamos variantes a partir de datos de secuencia este debe de distinguir entre mutaciones verdaderas y errores, lo cual es una tarea central del análisis genómico y, a menudo, requiere técnicas estadísticas, computacionales y/o heurísticas sofisticadas.
¿Por qué debo de elegir con cuidado la herramienta para el llamado de variantes?
Aunque los programas de llamado de variantes buscan superar el ruido inherente a los experimentos biológicos, su funcionamiento puede verse significativamente afectada por factores externos e internos, incluidos la biología de la muestra, las condiciones de los datos, el tipo de variantes que se deseen estudiar, el tipo de muestra e incluso cuestiones experimentales como el proceso de preparación, almacenamiento y análisis de muestras.
¿Es muy diverso el número de programas para el llamado de variantes?
Los llamadores de variantes son muy diversos en términos de sus algoritmos matemáticos centrales y entradas aceptables, los parámetros incluidos, el tipo de variantes que pueden identificar, así como características adicionales incluidas que deben de ser consideradas en el pipeline.
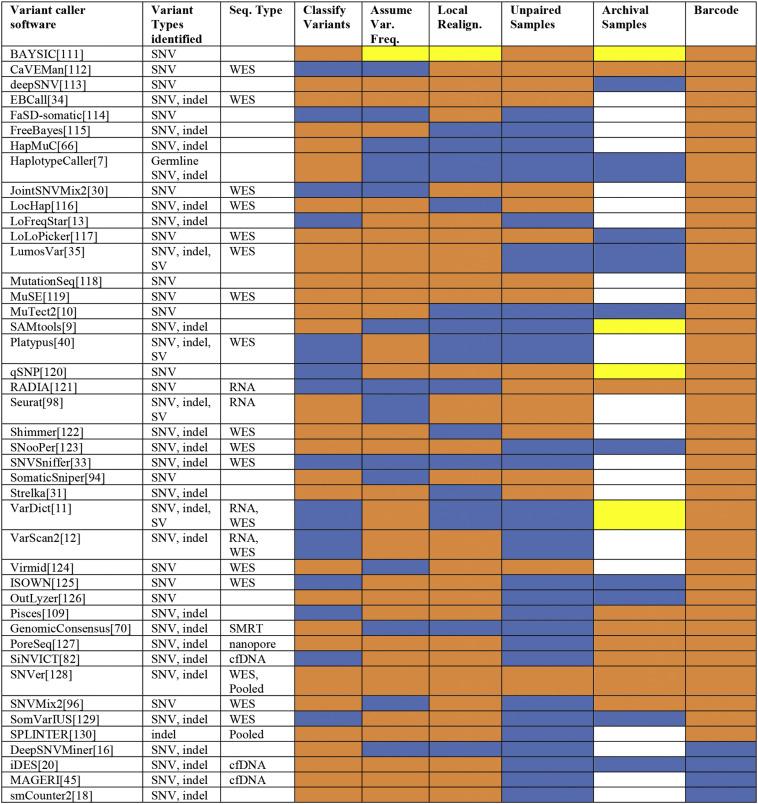
¿Son específicos para algún tipo de variantes?
- Línea germinal: HaplotypeCaller del Genome Analysis Tool Kit (GATK), MAQ y SAMtools mpilleup
- Línea somática (utilizados muy frecuentemente para cáncer): MuTect2, VarDict, VarScan2, LoFreqStar
¿Qué considerar?
El rendimiento de los llamadores de variantes puede verse afectado por factores de confusión experimentales, como la preparación de muestras, la preparación de bibliotecas y la tecnología de secuenciación.
Experimento previo a la secuenciación
- Muestreo y contaminación
- Conservación y Almacenamiento
- Preparación de la biblioteca
- Amplificación de la muestra
- Aislamiento de regiones de interés
- Identificadores de secuencias
Métodos de secuenciación
- Método basado en portaobjetos por síntesis (Illumina): Mutect2, LoFreqStar y JointSNVMix
- Método basada en microesferas por síntesis (Life Technologies/Ion Torrent): Llamantes variantes con opciones para establecer cortes de calidad de lectura como MuTect2 puede mostrar un rendimiento mejor o más confiable para los enfoques basados en perlas. Además, puede ser beneficioso seleccionar llamadores variantes que ejecuten la realineación local como parte de su método, como VarDict o HapMuC.
- Secuenciación en tiempo real de una sola molécula (Pacific Biosciences): Generalmente requieren algoritmos de llamada de variantes especializados, como GenomicConsensus distribuido por PacBio, y la alineación precisa y las variantes de llamada de estos datos son actualmente un área de investigación activa
- Secuenciación de nanoporos (Oxford): Esta tecnología tiene tasas de error relativamente altas y produce lecturas largas, por lo que requiere alineación especializada y algoritmos de llamada de variantes, incluido PoreSeq
Biología de la muestra
- Bajas frecuencias alélicas variantes: Dependiendo de diversos factores puede utilizarse DeepSNVMiner, smCounter2, o Platypus.
- Inestabilidad cromosómica: Además de pequeñas inserciones o deleciones (es decir, indeles) o mutaciones de un solo nucleótido, muchos cánceres se caracterizan por variantes estructurales (SV) más grandes, que pueden ser marcadores de la agresividad de la enfermedad y los resultados del tratamiento. Para la mayoría de los cánceres, las variantes en cada escala, desde la pérdida de cromosomas completos o brazos cromosómicos. Muchos programas de llamado de variantes se centran en la identificación de variantes relativamente pequeñas y no tienen en cuenta explícitamente alteraciones cromosómicas estructurales más grandes, que pueden ser mucho más difíciles de identificar sin algoritmos de identificación especializados. Algunos programas útiles: Seurat, Platypus y VarDic
Características clínicas
- Edad del paciente
- Heredabilidad
Fuente: Zachary S. Bohannan, Antonina Mitrofanova, Calling Variants in the Clinic: Informed Variant Calling Decisions Based on Biological, Clinical, and Laboratory Variables, Computational and Structural Biotechnology Journal, Volume 17, 2019, Pages 561-569,https://doi.org/10.1016/j.csbj.2019.04.002.

