
Las técnicas de secuenciación de alto rendimiento son capaces de producir millones de secuencias de nucleótidos del ADN de un individuo proporcionando una cobertura múltiple de genomas completos o regiones genómicas particulares. El resultado de la secuenciación de alto rendimiento consiste en conjuntos de secuencias genómicas relativamente cortas, generalmente denominadas lecturas. Las tecnologías de secuenciación contemporáneas son capaces de generar decenas de millones a miles de millones de lecturas por muestra, con longitudes de lectura que van desde unos pocos cientos hasta unos pocos millones de pares de bases el establecer las comparaciones y encontrar las diferencias entre el genoma de referencia y la nuestra muestra es lo que conocemos como alineación y mapeo.
¿Cuál es la diferencia entre alineamiento y mapeo?
Mapeo: Encuentra el origen aproximado de una secuencia. Es el proceso de establecer la relación espacial de los puntos de referencia dentro del ADN. Estos puntos de referencia pueden ser genes o regiones que varían entre individuos. El mapeo puede implicar simplemente ordenar dichos puntos o, en algunos casos, determinar con precisión el espacio entre ellos.

Alineación: encuentre la diferencia exacta entre dos secuencias.

Podríamos decir que el alineamiento es parte del mapeo el encontrar la secuencia de referencia y establecer una comparación para detectar las diferencias.
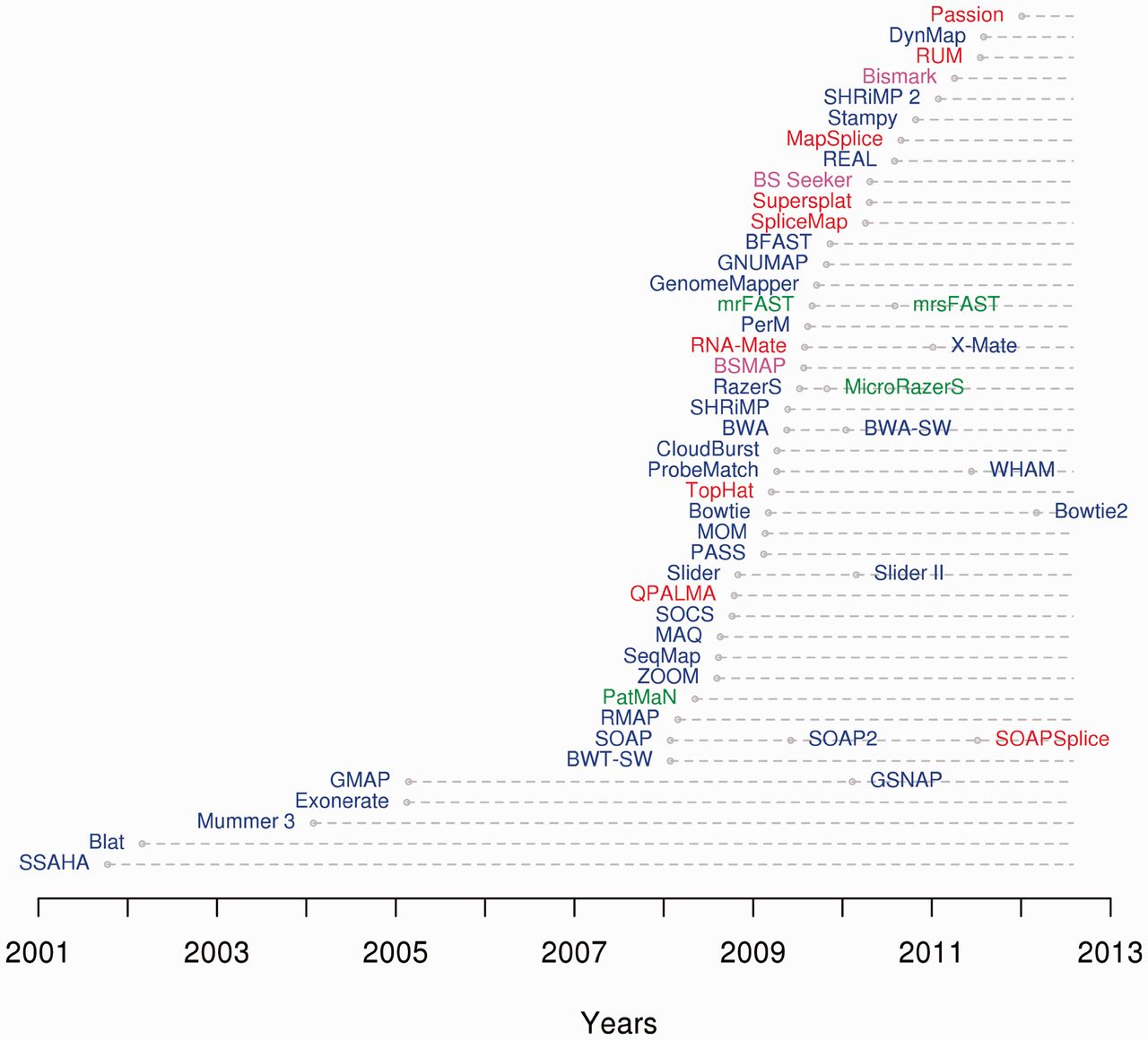
¿Cuáles son las herramientas más populares para alineamiento y mapeo?
El objetivo principal del mapeo y el alineamiento es encontrar la verdadera ubicación de cada secuencia a partir de una cantidad potencialmente grande de datos de referencia, teniendo en cuenta los errores y la variación estructural. Para permitir estos errores/variantes, la coincidencia debe ser aproximada. Las medidas de distancia que se utilizan normalmente tienen en cuenta la cantidad de desajustes e indeles para permitir errores y variaciones estructurales, pero también pueden incorporar tamaños de brecha o probabilidades asociadas con las lecturas. Actualmente, hay más de 60 mapeadores disponibles, la mayoría de ellos han sido propuestos después del 2008.
¿Qué consideraciones debo tomar en cuenta?
El primer paso en proceso directo y el análisis de los datos de NGS es la alineación y el mapeo. Se han propuesto numerosos alineadores que utilizan diversos algoritmos para estas tareas, hasta ahora todos cuentan con propiedades distintivas para lograr una mayor exactitud relacionada con la precisión.

Distintos estudios han revisado las herramientas según la estrategia de mapeo, las características de la herramienta, el tipo de datos de entrada y la manipulación de las opciones de parámetros de la herramienta. Por lo tanto, dentro de los que se debe considerar al seleccionar una herramienta de alineamiento:
El tipo de datos
Dependiendo de las muestras la herramienta adecuada para este proceso puede ser diferente esto está en función de las características propias de los datos o la muestra incluyendo si son datos de DNA (BWA), RNA (STAR,TopHat) , microRNA(STAR, BWA,SOAP,Bowtie1), etc.
La plataforma de secuenciación que generó los datos
Los avances en las tecnologías de secuenciación han permitido generar lecturas de más de 1000 bases dependiendo de la longitud de la lectura y su plataforma puede haber una herramienta ideal. Por ejemplo, BWA, Mosaik, SHRiMP y SOAP brindan resultados de mapeo satisfactorios en las alineaciones de lectura de Illumina SE y PE, también hay software que cada vez se vuelve más especifico para tratar con lecturas muy largas como BWA-SW O RUM.
Calidad en el mapeo
Considerar la calidad en el mapeo se vuelve fundamental dependiendo de la herramienta la eficiencia de este parámetro suele variar y es fundamental cuando se establece un umbral para filtrar las lecturas desalineadas de las lecturas correctamente alineadas. En alguno estudios se ha detectado que la distribución de calidad de mapeo para lecturas mapeadas correctamente en BWA-MEM en comparación con otras herramientas, más del 99 % de las lecturas mapeadas correctamente poseían una calidad de mapeo de más de 50. Otras herramientas como BWA-PSSM y Bowtie2 también han presentado valores altos.
Tiempo de ejecución y requisitos de memoria
Un mapeador útil debe poder equilibrar la velocidad y el uso de la memoria mientras informa las asignaciones deseadas. Dependiendo de este hecho se han hecho mútiples análisis donde se ha encontrado que dentro de este tipo de software BWA es de los que consume menor espacio de memoria, también se encontró que en cuestión de tiempo de ejecución herramientas como NovoAlign, BWA-MEM, Bowtie 2, BatAlign y BWA-PSSM son sumamente útiles pero quien mostro mejor rendimiento estable en multiples pruebas fue BWA-MEM.

¿A qué se deben las diferencias entre herramientas?
Las diferencias entre los métodos de mapeo y sensibilidades podrían atribuirse principalmente a las heurísticas aplicadas por diferentes algoritmos para mapear y alinear y detectar personas erróneas.
Fuentes
- Alser, M., Rotman, J., Deshpande, D. et al. Technology dictates algorithms: recent developments in read alignment. Genome Biol 22, 249 (2021). https://doi.org/10.1186/s13059-021-02443-7
- Lee, H., Lee, K. W., Lee, T., Park, D., Chung, J., Lee, C., Park, W. Y., & Son, D. S. (2018). Performance evaluation method for read mapping tool in clinical panel sequencing. Genes & genomics, 40(2), 189–197. https://doi.org/10.1007/s13258-017-0621-9
- Nuno A. Fonseca, Johan Rung, Alvis Brazma, John C. Marioni, Tools for mapping high-throughput sequencing data, Bioinformatics, Volume 28, Issue 24, December 2012, Pages 3169–3177, https://doi.org/10.1093/bioinformatics/bts60

